by John Hamlin
Editorial note: This article was written prior to the new FDA Guidance for Aseptic Processing being published. The second part of this article to be published in the near future will reflect the new Guidance recommendations.
For a more recent article by John Hamlin, please see “Introduction to Cleanrooms“.
Article Overview
Provide an overview of the critical manufacturing process, aseptic fill/finish production of sterile products.
This article is the first of a two part series to provide a broad overview of the aseptic fill/finish manufacturing process. This first article will discuss the background of aseptic products and the operational requirements of the aseptic operation. This will include the personnel, cleanroom, preparations, and the fill/finish process equipment and a brief discussion of the sterile lyophilzation requirements. The second article will discuss the global regulatory and compliance requirements and will include the process validation of an aseptic manufacturing operation.
Introduction
Aseptic filling of sterile drugs, also know as sterile filling, still remains one of the most critical processes in biopharmaceutical manufacturing. This is due to its highly technique driven processes and the potential safety impact to the end user, usually an already compromised patient. There are only indirect safeguards for the sterility of the filled drug after it is stoppered and capped in the cleanroom.
Unlike terminal sterilized filled drugs, the stability of the aseptic filled drugs will be affected by steam autoclave, dry heat ovens, Ethylene Oxide, and irradiation, either Cobalt 60 Gamma or E Beam. Thus the need to utilize an aseptic process to fill certain biologicals, pharmaceuticals and biotechnology drugs.
The history of aseptic fill/finish processing is relatively recent with the sterility requirements for injectables being established in the 1920s and large scale biological manufacturing of blood and plasma products during WWII. Plasma products did have, and some products still use, a post-fill pasteurization process of low heat treatment of 60°C for 10 hours. Pasteurization does not provide sterility, but can reduce the contamination of fungi. Anti-fungicidal reagents were also added to parenteral drugs to help mitigate the contamination that was occurring with early aseptic processing.
Then in an effort to help improve consistency in aseptic processing, the Parenteral Drug Association (PDA) published its Aseptic Validation Technical Report in 1981 [8]. This was followed by the Food & Drug Administration (FDA) in 1987 with its Aseptic Processing Guidelines [1]. The International Society of Pharmaceutical Engineering (ISPE) published its Sterile Facilities as part of their Guidelines Series in 1999 [14]. Recently, the FDA published its Concept Paper: Aseptic Guidelines in 2003 [15].
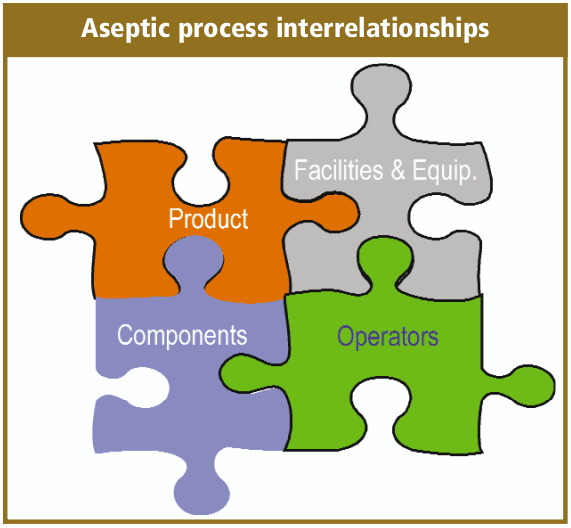
Aseptic filling is an aseptic process that requires the close coordination and complex interaction between personnel, sterilized product, the fill/finish equipment system, cleanroom and support facilities, and sterilized filling components.
There is also the perception issue for aseptic fill/finish, which is another reason for the many safeguards that I will discuss shortly, since micro contamination is not readily visible. Micro contamination is very small, and the surfaces that look clean and sterile may in fact not be. Thus the aseptic fill/finish processes are highly dependent on technique, detailed procedures, equipment and controls.
Regulatory Considerations
As with our industry, there are many global regulatory requirements for aseptic/ sterile fill/finish manufacturing. Although each country or geography has its regulatory guidance, we have not yet achieved full harmonization. Most of these are listed in this article’s appendix, and I will be only briefly discussing the current FDA 1987 Guidance. This FDA Guidance provides a couple of nice definitions for us.
“In aseptic processing, the drug product, container and closure are subjected to sterilization processes separately and then brought together Because there is no further processing to sterilize the product after it is in its final container; it is critical to the maintenance of product sterility that containers be filled and closed in an environment of extremelv high quality”
We also have written in the Code of Federal Regulation (CFR), section 21 CFR 211.113 (b) that states
“Appropriate written procedures, designed to prevent microbiological contamination of drug products purporting to be sterile, shall be established and followed. Such procedures shall include validation of any sterilization processes.”
Another section, 21 CFR 211.167 (a) states
“For each batch of drug product purporting to be sterile and/or pyrogen-free, there shall be appropriate laboratory testing to determine conformance to such requirements. The test procedure shall be in writing and shall be followed.”
Currently, the FDA has been expressing a number of concerns about aseptic manufacturing, citing all drugs recalled due to non-sterility over the last 10 years were produced by aseptic processing (Spring 2002). If you drill down in these recalls, you will find that there are a few companies who have multiple recalls, and that there are a lot of “documentation” recalls. These are situations in which the documentation or procedures had omissions and errors and as a result a recall was initiated. The consensus within our industry is that, in fact, we have been getting much better with our aseptic filling processes
Aseptic Fill/Finish

What can be aseptically filled? Virtually any solution, powder or suspension that can be terminally sterilized prior to the aseptic fill/finish process. Typically sterile drugs are aseptic fill/finish in molded glass bottles, tubular glass vials, tubular glass syringes and in Europe more than the United States, glass ampoules. Solutions can also be subsequently lyophilized in a sterile dryer to further stabilize drugs. The more unique the product or container system, the greater the technical or operational challenges that may ensue.
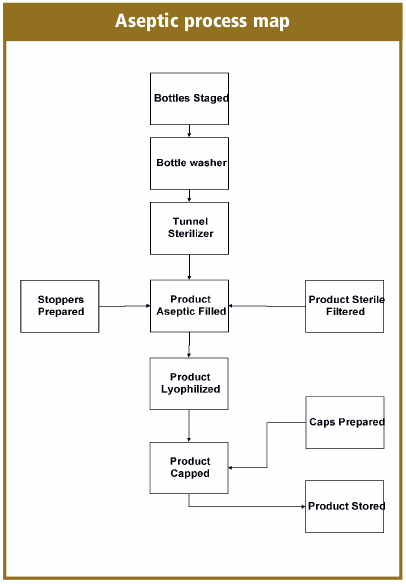
How do we complete the aseptic fill/finish process? You need to decontaminate the operational personnel, terminally sterilize the drug product, filling components, equipment change parts and sanitize the cleanroom and in-place equipment. Then bring it all together with good aseptic practices, and the simplified process maps look like the aseptic process map.
The aseptic fill/finish methods can vary between an early clinical phase hand fill (clinical solution fill photo), to small volume semi-automated filling to the fully automated high-volume over multiple day production batches.
Cleanroom Personnel
The personnel in the cleanroom are like the “double-edged” sword, they are absolutely necessary to complete the aseptic fill/finish process, but at the same time, provide the greatest microbial risk for a sterile product. You are constantly regenerating yourself, and in the process shedding a huge amount of dead skin and other particles. An average person is capable of shedding ten million particles per day. That is further compounded by the types of clothes worn and where you have recently been, such as what might be on the bottom of your shoes right now.
Thus the amount of investment that is made to limit the operational personnel risk to the sterile drug. Personnel are encapsulated with extensive sterile gowns and qualified gowning procedures. The cleanrooms have extensive unidirectional flow air currents to provide a barrier as well as sweep the potential contamination away from the exposed drugs.
Automated fill/ finish equipment is installed to reduce the amount of personnel who are present in the cleanroom during the aseptic fill/finish processing. Development of the current barrier equipment designs and the recent development of the isolator technology have been made to further isolate the exposed sterile drug from the operational personnel.
Lastly, the implementation of Best Aseptic Practices to provide personnel with methods, training and qualified procedures to further prevent microbial contamination of the sterile drugs.
The Best Aseptic Practices are a set of best practice methods for personnel to govem themselves as they move and function in the cleanroom environment while executing their processes. Aseptic practices will also incorporate the specific requirements for your aseptic fill/finish processing. Procedures are needed for both gowning and de-gowning processes, and state hygiene, training, qualification and re-qualification requirements.
Sterile outer garments are usually made of synthetic or natural materials, worn as an outer garment, which have low or no particle shedding or penetration characteristics. Most companies outsource their sterile garment preparation to a company who will wash and sterilize their garments, usually sterilize with Gamma. For low volume sterile garmenting requirements, you can utilize single-use sterile garment packs. The sterile outer garments act as a personnel filter to isolate the individual and their contaminants from the cleanroom environment and the sterile drugs.
Personnel who function in the aseptic fill/finish aseptic processing core will need to have completed a gowning qualification, especially to be present in the clean room core during a sterile fill operation. This would include the operational personnel, maintenance mechanics, quality assurance and quality control personnel, production management, engineers and technicians. The qualification should include training on the basics of microbiology and the Best Aseptic Practices. Typically, this is followed by a gowning demonstration, then a gowning critique of the person in training.
Final gowning qualification should be completed with multiple sterile gowning in the cleanroom with microbial testing inside the cleanroom. I recommend that the sterile gowning and microbial events should be videotaped to provide the operator with additional feedback and assist with the analysis of the gowning techniques. Gown qualification best practices require the gowning qualification to pass three consecutive microbial testing and successful media participation prior to being deemed gowning qualified. An example of a gowning process is provided in Table 2.
Facilities: The Cleanroom
The cleanrooms are controlled areas and in conjunction with the supporting utility systems and facility infrastructure, create the environmental envelop in which the aseptic fill/finish process operates. As with the other components of the aseptic processing, the cleanrooms area complex combination of physical rooms and areas, utilizing High Efficiency Particulate Air (HEPA) to create unidirectional air patterns, maintenance of positive pressure between rooms in conjunction with constant air changes, and sanitization processes. All of this operates with constant environmental monitoring (EM).
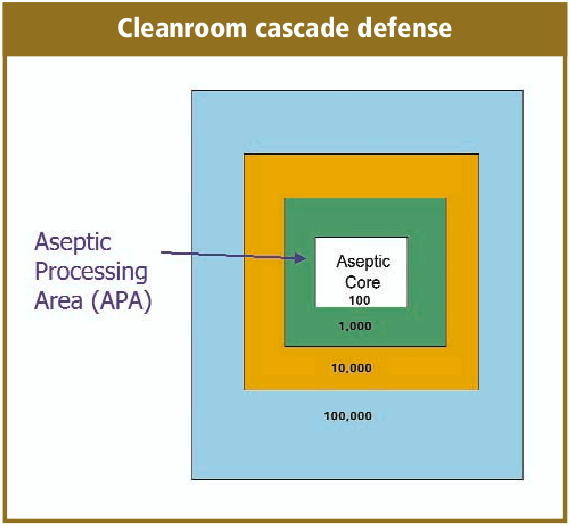
The cleanroom design will take into consideration the flow of personnel, product, equipment and components. Effective aseptic fill/ finish facility designs will take into account the flow of all of these from the receipt of raw materials at the warehouse through the facility to final warehousing. A very simplified illustration is the cleanroom cascade defense.
Very basic to the cleanroom design is the classification of the cleanrooms relative to the operation that is occurring within it as, well as adjacent to it. Harmonization of the regulatory guidelines for cleanrooms has not fully occurred yet, but I believe we are making some progress. In the cleanroom classification table (Table 3) is a very simplified comparison between the European Annex l and FDA classifications. I have referenced the various cleanroom compliance documents in the article appendix, and an in-depth discussion of cleanroom classifications was not intended for this article. You will need to know where your products are going to be distributed to select the proper guidance to follow, which for our industry and global products, usually means all of them.
Before discussing the cleanroom materials of construction or the Heating, Ventilation and Air Condition (HVAC), it is critical to first understand the flow of personnel, sterilized components and sterile product in developing the cleanroom design and operation. The flow requirements may vary with each sterile drug produced.
The personnel flow, as I discussed earlier, is very critical to maintaining the sterile environment. This would include the gowning, degowning and all of the necessary movements through all of the cleanroom facilities. It is ideal to ensure that the personnel flow is moving one-way; from gowning to operation and then degowning, cleanest area towards the “dirtiest.”
The one-way movement within the cleanroom, especially the sterile core for the aseptic fill/finish operation, is critical for all of the material, products and components. You will want to ensure your cleanroom design will eliminate two-way transfers from occurring concurrently, where sterile goods are physically passing “non-sterile” goods where there is a potential for microbial cross-contamination.
The movement of mobile tanks with sterile filter bulk drug presents challenges as well, as the exterior surfaces cannot be terminally sterilized with the drug enclosed before the aseptic fill/finish operation. The bulk tanks will require sanitization in airlocks or at other transfer modules. The sanitization processes for mobile tanks are challenged by the amount of fixtures on the tanks, clearance under the tank, and the tank wheel assemblies. Frequently the mobile tanks are segregated from the aseptic core and only the transfer of the bulk tank tubing connection necessary for the aseptic connection.
The equipment layout and flow will also influence the cleanroom design. The ideal aseptic fill/ finish system is a fully automated in-line isolator fill/finish system. The most difficult to manage and presenting the greater microbial risk, is a batch sterilization and completely manual filling process that occurs in a bio-hazard safety hood.
The equipment flow should also reflect the necessary sterilized set-up parts that will be changed for each sterile drug batch, such as the filling needles, stopper bowl and feeder components. The sterilized set-up components may require a specialized technician or mechanic to support the aseptic fill/finish operation. The ease in which the aseptic set-up can be accomplished and maintained can directly influence the quality of the aseptic fill/finish operation. You should eliminate any operations that require a sterile operator to reach over the fill line.
The aseptic core in which the sterile drug is actually exposed to the cleanroom environment is the most crucial area of a cleanroom, and warrants the most detailed attention to the design of the cleanroom. This is the area where the sterile drug is transferred from the filling needles to the sterile container. Typically the stoppering or closing of the container occurs immediately after, with the exception of when the drug requires sterile lyophilization. The requirements of the lyophilization process require the stopper be only partially seated on the vial.
For solution drugs after a stoppering process, sealing occurs immediately, usually with some kind of aluminium seal. The design of the cleanroom or equipment would include a barrier between the stoppering and sealing processes to minimize any potential aluminium contamination.
Another variation for sterile drug solutions is the use of Form-Fill-Seal (F-F-S). This fully automated process forms a plastic container system, filling the solution during the process and immediately sealing the containers. The F-F-S process minimizes the environmental exposure and provides microbial contamination results similar to an isolator process.
For lyophilized drugs, the filled and partially stoppered vials would be transferred to a sterile lyophilizer (drier) for the completion of the lyophilization cycle. It is normal for the stoppers to be seated in the vials inside the sterile drier at the end of the lyophilization cycle prior to opening the door. The stoppered vials are then removed from the sterile drier and immediately capped. The delay in sealing the container, immediately after the filling process, allows the drug to be exposed to the environment is an additional risk that occurs with sterile lyophilization.
An essential component to the cleanroom is the Heating, Ventilation and Air Condition (HVAC) systems. The HVAC systems that support pharmaceutical operations, especially cleanrooms and aseptic manufacturing, are complex and extensive. The heating and cooling functions are needed for operator comfort and environmental control. Ventilation function provides the necessary circulation and “air turns” to maintain environmental control. The HVAC will also be designed and operated to maintain the aseptic core by the use of positive pressure that extends away from the core.
A rule of thumb for temperature in the heavily gowned aseptic core is mid 60°F, and the relative humidity above 30% to control static, and below 60% to control rust, organism growth and personnel from sweating.
For cleanrooms, the ducts terminate at High Efficiency Particulate Air (HEPA) filters. For the HVAC that supports the aseptic processing operations, including the preparations area, there will be a series of pre-filters prior to the HEPA filters. The HEPA filters are rated 99.95% effective for microbial retention and facilitate unidirectional air flow. Previously, it was thought that a laminar air flow pattern could be effectively achieved with the HEPA filters, but with the knowledge gained by extensive smoke studies of class 100 aseptic cores, the more realistic expectation is a unidirectional air flow pattern.
The HEPA filters are the achilles heel of the cleanroom HVAC system. They require extensive care and maintenance and could have a detrimental effect on the quality of the cleanroom environment if not well maintained. HEPA filters have the potential to fail within the filter medium, at the gaskets, seals and frame.

Other utilities that are needed to support the aseptic fill/finish operation include Water for Injection (WFI), oil-less compressed air, nitrogen gas, sterile steam and vacuum. The compressed air and nitrogen gas will also have point of use sterile filters inside the aseptic core, and the vacuum system should have one-way check valves. The WF I is predominately used in the preparations for the rinsing of vials, stoppers and equipment change parts. The intent of this article was not to provide an overview of the utility design and operation that support cleanroom operations.
Materials of construction of a cleanroom should facilitate the required operation, which includes extensive cleaning processes and support the required environment control. The surfaces should be hard, smooth and easily cleanable. The floors, ceiling and walls should be continuous, with flush installations and utilizing welded joints where possible. The wall fixtures should be flush mounted to surfaces and the lighting fixtures flush mounted and preferably remote access. Surfaces should not be designed to allow the build up of particulate contamination.
Most aseptic cleanrooms have telecommunication equipment to allow discussions without the need of personnel leaving and entering the operation. Increasingly, video monitoring and recording cameras are installed in the aseptic core. The video equipment allows a further reduction of monitoring personnel inside the critical area, where each additional person incrementally increases the risk of microbial contamination.
Cleanroom maintenance and sanitization requires the qualification of the cleaning and disinfectants agents. The qualification of the sanitization processes will need to be done in conjunction with a documented process and trained personnel. This qualification should include the development of the expiration dates for the formulated sanitization solutions.
Some of the common disinfectants and sterilants are phenolics, sterile alcohol, hydrogen peroxide, Quaternary Ammonium, Sodium Hypochlorite, and Formalin. Phenolics will work with organic matter, but have issues with resistant spores. Alcohols will act quickly, but have little effect on spores and are flammable (e.g. 70% Isopropyl Alcohol, IPA). Hydrogen Peroxide is an excellent sporicidal, but also not compatible with all surface agents (e.g. “soap”). Quaternary Ammonium (Quarts) are highly stable and nontoxic, but affected by water quality, soap, and not effective with spores. Formalin is effective against bacteria spores, will not corrode metal, but it is toxic with irritating fumes and residuals that can be difficult to clean. Sodium Hypochlorite is an excellent sporicidal but affects metals.
The cleanroom sanitization process requires full sterile gowning and all of the required aseptic techniques that would be utilized during the aseptic filling. As with the aseptic filling process, the cleanroom sanitization process requires documentation, personnel training and qualification. Environmental Monitoring (EM) is the process to ensure that the cleanroom is under control for potential viable and non-viable contamination. Your EM process should have qualified methodologies to routinely collect, evaluate and interpret EM data. The determination of sampling points and required limits should be defined in your documentation. Your EM program should identify periods of critical activity where sterile product may be exposed to environmental conditions (photo Em class 100 Bio Safety Hood).
Sterile Product Filtration
Your drug will require sterilization by some method prior to the aseptic filling process. Traditionally, the bulk drug sterilization is accomplished by filtration, normally a depth filter. You will need to bulk drug a method for sterilization and a sterile container system that is compatible with the drug and your aseptic fill/finish process. The drugs can be pre-sterile filtered (e.g. .45 micron), followed by a series of at least two sterile filters at .22 micron. The sterile filters are both pre- and post-bubble tested to ensure integrity. The Sterile bulk is then transferred to the aseptic fill and aseptically connected to the fill equipment. Currently the best-in-class for sterile filtration is a closed system that extends from the non-sterile bulk to the aseptic filling equipment.
Filling Components

All components and supplies that are required during the aseptic fill/finish operation must be either sterilized or sanitized. Sterilization is usually completed with pass-through steam autoclaves, dry-heat oven or tunnel and sanitized cleanroom airlocks.
The filling components can also vary per the drug and production requirements. Tubular Type I glass vials are predominately used for Small Volume Parenterals (SVP) and sterile lyophilization and blown Type II glass bottles for Large Volume Parenterals (LVP). Tubular Type I glass stock is also predominately used for aseptic syringe production. A number of manufacturers are considering Cyclic Olefin Copolymer (COC) vials that function similar to glass vials.
The stoppering of the vial provides the sterile seal of the drug from the environment and a crimp-seal cap ensures the long term integrity. The stopper also provides a barrier to gas and oxygen to the drug ensuring long term stability. Elastomeric closures (stoppers) that are used for parenteral solutions are formulated to ensure product stability and patient functionality. Two of the basic styles of closures are the “plug” for sterile solutions and the “leg” for sterile lyophilization (clinical solution fill photo). Some of the considerations should be given to size, type and number of needle punctures, water vapor transmission rate, ability to retain bound water, gas transmission, stoppering equipment of the filling line and potential extractables.
Component Preparation
There are many ways to aseptically fill/finish sterile drugs, which includes the traditional solution filling of glass vials and syringes, sterile powder fills, sterile lyophilization, and blow-fill-seal. For this article, I will only be addressing solution filling and sterile lyophilization. There are many considerations in the selection of your aseptic filling equipment. To name but a few: solution volume, fill tolerance, production throughput, drug viscosity, drug foaming, gas blanketing, drug temperature, potent compounds, drug stability and reactivity.
Sterile preparation of the vials and bottles is achieved by rinsing (washing) to remove endotoxins. The glass vials and bottles are depyrogenation usually with hot air. This is accomplished in a batch mode with an oven, or a continuous process with a tunnel that connects the bottle washer to the filling station.
For small parts cleaning, such as filling needles, forceps and stoppering equipment, as well as stoppers, you will complete the initial washing/rinsing to remove endotoxins and loose particulate. Then wrap the parts for subsequent steam autoclave processing to destroy the endotoxins. Depending on the formulation, the stoppers may be able to be sterilized by irradiation.
Aseptic fill/finish processes can very from a clinical hand fill, to semi-automated mono-block, to a high speed filling lines. Filling equipment systems can be characterized as either “Open,” “Barrier,” “Isolator” and “RABS.”
Filling lines are characterized as having no barriers or other physical restrictions between the sterile operator and the sterile drugs. As a result of EU regulation, open fill lines are not common to commercial aseptic operation, but can be found in Phase I and II clinical manufacturing operations. The barrier filling lines have transparent panels that restrict sterile operator access to the sterile drug. Some of the barrier panels may be designed as doors to the Barrier with very specific operational procedures that support aseptic techniques for use during aseptic fill/finish production.
The solution filling process will include the transport of sterilized vials and bottles, orientation to the filling station, a means for check weighing, stoppering and crimping stations. For high speed lines, there will also be accumulation tables and vial load/loading stations. The filling equipment can include the sophistication of in-line check weigher, automated vision systems, reject stations, and SCADA information systems networked from each equipment’s PLC.
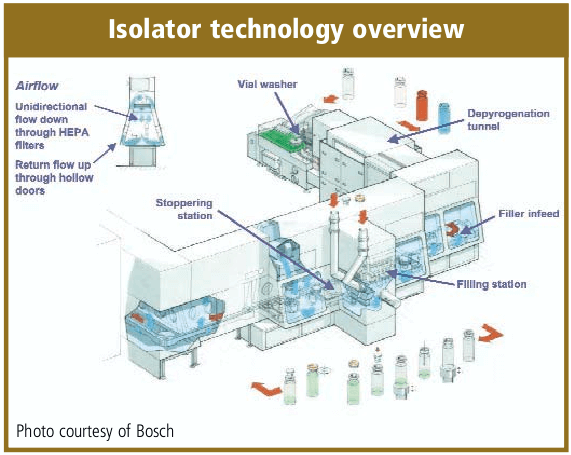
As a rule of thumb, the stoppering and capping (sealing) should be completed as soon as possible. There is some discussion that the crimping of the cap may not require the same critical environment as the solution filling process and crimping may be a particulate generating process. The norm for solution filling equipment is to provide a barrier between the stoppering and capping processes. Isolator systems are a current alternative to the classic barrier equipment installation. Isolators utilize a glove box technology and they are designed for minimal human intervention which provides increased contamination control. A majority of the isolators are sanitized by vaporized hydrogen peroxide. Isolators require more expensive capital investment, can be more complex to install, qualify and operate and may have less flexibility to changeover fill sizes and products. They have historically been designed for high-volume dedicated drug production and microbiological quality laboratory operations. There is also a trend to utilize Campaigning for Isolators technology installations [16].
An alternative to isolator technology is the “Restricted Access Barrier System” (RABS) a term first described by Upjohn, now Pfizer. RABS is similar to the isolator technology utilizing glove ports and other sterile operator restrictions. It is also similar to the traditional barrier fill line with the utilization of a conventional aseptic core cleanroom. The gowning and aseptic techniques are the same as a barrier fill/finish operation. The advantages that have been reported are reduced capital investment, quicker validations and operational start-up, reduction in lot-to-lot turn around time. RABS operations have documented contamination control over a traditional barrier fill/finish system.
Sterile Lyophilization

A sterile lyophilization process requires all of the basics for aseptic processing of a solution product, but with the additional processing requirements and risks of the sterile dryer (Lyo) equipment. Sterile dryers are now designed to utilize Clean-in-Place (CIP) and Sterilization-in-Place (SIP) of both the condenser and the product chamber.
The chamber which holds the drug product being processed requires a loading methodology that is consistent with aseptic techniques. For high production and large sterile dryers, the majority of new installations also include automated load and unload equipment. The automated load/unload capability reduces the headcount inside the aseptic core and should reduce the risk to microbial contamination.
The lyophilization process includes filling the product solution aseptically, with the stopper partially seated in the vial. The partially stoppered vial is then transported and loaded into the sterile dryer, thus the sterile product has an extended exposure to the environment. The drug solution is then frozen by either immersion in liquid nitrogen prior to loading or by the sterile shelf. The lyophilization cycle includes the primary and secondary (terminal) drying. After the lyophilization cycle has been completed, the stoppers are usually seated into the vial by lowering the dryer shelves. A sterile drug producer may need to stopper the lyophilized vials under vacuum or and inert gas. Then the dryer door is opened and the stoppered vials are transported to a capping (crimping) process.
Media fills for process validation for a sterile dryer is not a full process simulation. The lyophilization process is usually conducted under near vacuum, with a slight amount of pressure provided by sterile nitrogen and at -35°C or colder. All three of these variables have a negative effect on media and will distort the results. Thus, most companies will modify the media fill in the sterile dryer by not freezing the shelves, not evacuating the chamber and connecting sterile air to the chamber inlet.
Visual Packaging Inspection
Visual packaging inspection of aseptic filled drugs is usually completed 14 days after fill. This is a period of time that could allow the growth of any potential contaminating micro organisms. The critical inspection process is for the presence of a cloudy or hazy solution that would indicate a contamination potential. The manual version of this inspection occurs with the use of white and black background viewing areas.
Manual visual inspection requires trained and tested inspectors, and due to the repetition of the inspection task, it is really only about 85% effective. Thus a number of companies have implemented double- inspection of the product, very tight acceptance criteria and automation of the process with vision systems.
Appendix
- Guideline on Sterile Drug Products Produced by Aseptic Processes, FDA, pub. 1987.
- Guidance for Submitting Documentation for Sterilization Process Validation in applications for Human and Veterinary Drug Products, FDA, pub. 1993.
- ISO 13408-1 Aseptic Processing of Health Care Products.
- ISO 14644 Cleanroom Standard (replaces MIL-STD 209E, obsolete 2001).
- Annex 1, Manufacturer of Sterile Medicinal Products, Pharmaceutical Inspection Convention, pub. 2003.
- USP <61> Microbial Limits.
- USP <1116> Cleanrooms.
- Validation of Aseptic Filling for Solution Drug Products, PDA Technical Monogram, Number 2, pub. 1981.
- Process Simulation Testing for Aseptically Filled Products, PDA Technical Report Number 22, pub. 1996.
- Points to Consider for Aseptic Processing, PDA, pub. 2003.
- Current practices in the Validation of Aseptic Processing, PDA Technical Report Number 36, pub. 2002.
- Design & Validation of Isolator Systems for the Manufacturer and Testing of Health Care Products, PDA, Technical Report Number 34, pub. 2001.
- Aseptic Pharmaceutical Manufacturing, published 1987 by Interpharm Press.
- ISPE Baseline Pharmaceutical Engineering Guide, Volume 3, Sterile Manufacturing Facilities, Jan. 1999.
- FDA Concept Paper on Aseptic Processing: www.fda.gov/cder/dmpq/aseptic-cp.pdf
- D. Stockdale, “Isolator Campaigning – An Industry Survey & Discussion of Operational Practices ”, published by ISPE Pharmaceutical Engineering Magazine, 2004.
Table 1. Definition of Terms for Aseptic ProcessingBasic terms defined
(source: ISPE Definition of terms) |
Table 2. Basic Aseptic Cleanroom Gowning
Note: Gowning in Critical area (Class 100/Grade A) becomes more stringent when working with exposed sterile drugs.Operators should gown with additional sterile sleeves, sterile glove changes, & sanitize of gloves. |
Table 3. Cleanroom cIassification examples |
|
| FDA categories | EU annex 1: 4 categories (A-D) |
| Class 100 | GradeA (Critical 100) |
| Class 1,000 (optional) | Grade B (Aseptic 100 non-unidirectional) |
| Class 10,000 | Grade C (Controlled l0,000) |
| Class 100,000 | Grade D (l00,000) |
| Douglas Stockdale is the President of Stockdale Associates, Inc., which provides extensive aseptic fill/finish and sterile packaging consulting services for the life sciences industry. He had twenty years of operational experience with Baxter Healthcare prior to founding Stockdale Associates. He is an internationally known expert consultant, speaken and writer about the issues of aseptic fill/finish and sterile packaging. His company provides innovative strategic and implementation solutions for product development, manufacturing, compliance/auditing and validation. He is a member of PDA, AAPS IOPP & ISPE and a member ofthe Board of Directors for the Greater Los Angeles chapter of ISPE. Mr Stockdale has a MBA jrom University of La Warne and B.S. in Engineering from Michigan State University. |
| Contacting the author: Douglas Stockdale, President, Stockdale Associates, Inc. Corporate Offices: 10 Reata, Rancho Santa Margarita, CA 92688, Phone: 949-888-9488, Fax: 949-888-1560 douglas@stockdale-inc.com or wwwstockdale-inc.com |
This article was printed in the September/October 2004 issue of American Pharmaceutical Review. Copyright rests with the publisher. For more information about APR and to read similar articles, visit www.americanpharmaceuticalreview.com and subscribe for free.